Hereditary retinal dystrophies (retinitis pigmentosa, Leber congenital amaurosis, cone-rod dystrophies, macular degeneration) are characterized by loss of visual function, sometimes starting during early childhood, other times in late adulthood. About 30% of these dystrophies are inherited in an autosomal dominant fashion (RetNet), caused by gain-of-function mutant alleles which encode a malignant form of a normal protein.
Dominant retinitis pigmentosa and macular degeneration are regarded as ideal targets in the development of gene therapies, as the eye is easily accessible for intervention. One way to cure dominant disease is to restrict production of the malignant protein in a safe and targeted manner. Allele-specific RNA interference (RNAi) is one ‘gene silencing’ therapeutic strategy. Promising results were reported in clinical and preclinical studies on dominantly inherited amyotrophic lateral sclerosis, spinocerebellar ataxia, Huntington’s disease and other nucleotide repeat diseases using small interfering (si)RNA and short hairpin (sh)RNA. RNAi studies not requiring resistant replacement include knockdown of vascular endothelial growth factor (VEGF) and its receptor in wet age-related macular degeneration (AMD). However, absent specificity and due to side effects, some clinical studies have been terminated (e. g., bevasiranip, a synthetic siRNA to treat macular degeneration; see also Bevasiranib terminated).
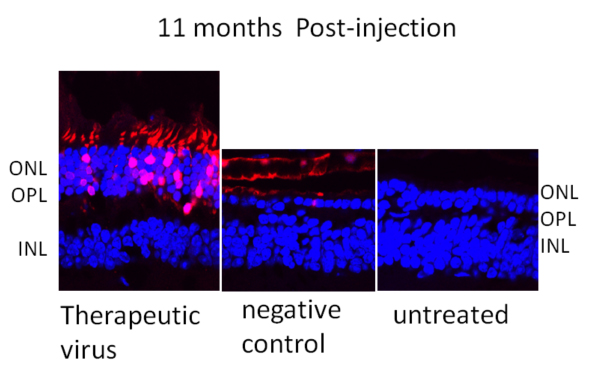
This study by Li Jiang, Houbin Zhang, Alexander M. Dizhoor, Shannon E. Boye, William W. Hauswirth, Jeanne M. Frederick and Wolfgang Baehr tested the therapeutic efficacy of RNA interference in two transgenic mouse lines that simulate dominant rod/cone dystrophy in severe (line L53) and moderate (line L52H) retinal degeneration forms. Inactivated viral particles were used to deliver the therapy. They generated viral (adeno-associated virus or AAV) particles that express RNA interference degrading specifically the malignant RNA. Viral particles were then injected into the subretinal space of L52H mice expressing a Y99C mutant bovine GCAP1 at three weeks of age and followed for one year. Encouragingly, ~80% knockdown of the mutant allele was achieved. This provided a significant, long-term improvement in retinal morphology, photoreceptor function (ERG) and behavioral measurements. Sustained preservation of retina morphology and function in this study suggested little, if any, off-target shRNA interference that is usually accompanied by increased cell death, or inactivation of the virus by induction of the antiviral interferon pathway. Importantly, the knockdown strategy is applicable to all GCAP1 mutations provided that the guide strand of shRNA is located outside hands EF3 and EF4 that contain the disease-causing mutations.
Studies like this offer hope for a cure of dominant cone dystrophy in human patients. The GCAP1 malignant allele in human patients would likely differ from its wildtype counterpart by a single base substitution. Therefore, an allele-specific shRNA degrading the mutant RNA cannot be generated. However, a nonallele-specific GCAP1 shRNA may be used to target both malignant and wildtype Guca1a alleles in a practical RNAi gene therapy approach. Since deletion of the GCAP genes in mouse had no detectable retina degeneration consequence, knockdown of both dominant mutant and wildtype Guca1a RNA species may suppress the human disease phenotype without pathology.